La cholangite sclérosante primitive (CSP) est une maladie rare, chronique du foie, caractérisée par une atteinte inflammatoire et fibrosante des voies biliaires, intra et/ou extra hépatiques. L’évolution de la maladie est variable et marquée par la formation de rétrécissements des canaux biliaires entravant l’écoulement de la bile. Elle est fréquemment associée (50% à 75% des cas) à une maladie inflammatoire de l’intestin. Elle touche des personnes souvent jeunes (âge habituellement inférieur à 40 ans au moment du diagnostic), plutôt des hommes (2/3 des cas). Contrairement à la cholangite biliaire primitive (CBP), cette maladie peut atteindre l’enfant.
Cet article a été rédigé en priorité à l’attention des patients. Pour des informations plutôt à destination du corps médical, nous vous invitons à visiter la page
Interview du Pr. Vincent Leroy sur Cholangite Sclérosante Primitive
(version courte 7mn – voir la version longue en bas de page)
Pour bien comprendre la CSP, il faut comprendre le rôle du foie et de la bile
Le foie est un organe de grande taille. Il représente à peu près 2 % de votre poids et pèse en moyenne 1,5 kg. Il se divise en 2 parties : le lobe droit qui représente environ 75 % de son volume total, et le lobe gauche, plus petit, donc. Il est placé sous les poumons, la partie la plus importante étant positionnée du côté droit.
C’est un organe fortement irrigué par le sang puisqu’il contient à chaque instant 10 à 15 % du volume sanguintotal de votre corps. Chaque minute, votre foie draine 1,5 litre de sang. Ce débit sanguin hépatique augmente lors du repas puis diminue lorsque vous dormez.

Les cellules du foie
Le foie est constitué de différents types de cellules dont :
- les hépatocytes principalement, qui ont un rôle essentiel dans la fabrication et la transformation des sucres, des graisses et des protéines, ainsi que dans l’élimination de substances étrangères, parfois toxiques (voir page suivante)
- les cholangiocytes, cellules participant à la sécrétion de la bile et tapissant les canaux qui la transportent à l’intérieur puis à l’extérieur du foie
- des cellules du système de défense naturel (système immunitaire)
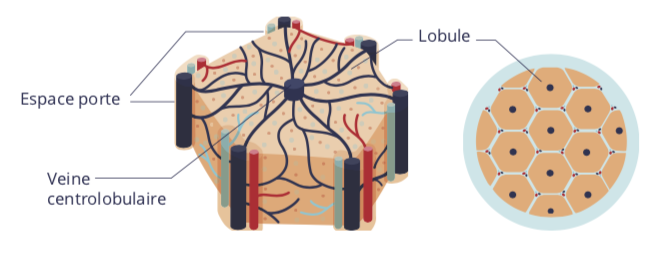
Les cellules du foie sont organisées en lobules. Ces lobules sont schématiquement hexagonaux et centrés autour d’une veine, dite « centrolobulaire ». Entre les lobules se trouvent les espaces portes. Ils participent à l’irrigation sanguine des lobules et à l’excrétion des éléments synthétisés par les cellules du foie, notamment la bile. Pour ce faire, ces espaces comportent une ou deux branche(s) artérielle(s), une branche veineuse et un ou deux conduit(s) biliaire(s), bordé(s) par les cholangiocytes.
Une aide précieuse pour le système digestif
Le foie sécrète environ 0,6 litre de bile chaque jour. Elle est produite dans le foie par les hépatocytes (75 %) et les cholangiocytes (25 %). Ce liquide d’aspect jaune-vert participe activement à la digestion.
La bile est collectée dans le foie par les voies biliaires dites intra-hépatiques puis circule en dehors du foie par les voies biliaires dites extra-hépatiques. Elle est stockée dans la vésicule biliaire à jeûn puis est libérée dans le tube digestif avant et après les repas.
La bile est composée principalement d’eau (97 %) et d’acides biliaires. Ces derniers facilitent la digestion et l’absorption des nutriments dans le tube digestif. Ils permettent notamment de rendre solubles les graisses d’origine alimentaire ainsi que toutes les substances qui ne se dissolvent que dans des graisses (molécules dites « liposolubles ») comme les vitamines A, D, E et K, certains médicaments, … Ainsi, la bile augmente le passage de ces nutriments dans la circulation sanguine.
Elle favorise aussi la sécrétion de molécules antimicrobiennes par l’intestin.
Qu’est-ce que la cholangite sclérosante primitive ?
Une atteinte des canaux biliaires
La cholangite sclérosante primitive (CSP) est une maladie du foie de longue durée, dite chronique. Elle se caractérise par une inflammation, un rétrécissement puis une obstruction des voies biliaires intra et extra-hépatiques par du tissu fibreux non fonctionnel (fibrose). La fibrose provoque une diminution, voire un arrêt, de l’écoulement de la bile, appelée cholestase chronique.
Que signifie « primitive » ?
La cholangite sclérosante est dite « primitive » lorsqu’aucune cause particulière n’est identifiée. Elle s’oppose aux cholangites sclérosantes dites « secondaires » qui peuvent être provoquées par des infections bactériennes répétées, une insuffisance d’apport en oxygène (ischémie) après une transplantation du foie par exemple, des substances toxiques comme certains médicaments, etc.
Des causes encore mal connues
La CSP est une maladie pour laquelle on suspecte en partie un mécanisme auto-immun. Une maladie auto-immune est une maladie liée à un dysfonctionnement de votre système de défenses naturelles qui se conduit de façon anormale et s’attaque à des constituants de votre organisme qu’il ne reconnait pas comme vous étant propres.
Mais d’autres phénomènes semblent aussi impliqués dans l’origine de son développement : épisodes infectieux, terrain génétique favorable, composition anormale de la bile, métabolisme anormal de la bile par la flore intestinale, etc. La CSP serait donc une réponse inflammatoire inadaptée des canaux biliaires à ces différentes agressions chez des personnes présentant une susceptibilité particulière d’ordre génétique.
Qui est concerné par la CSP ?

La CSP touche plus particulièrement les personnes jeunes. Elle peut parfois apparaître dès l’enfance.
Sa prévalence, c’est-à-dire le nombre de cas dans la population à un instant donné, n’est pas précisément connue. Les estimations montrent qu’il s’agit d’une maladie rare observée chez 6 à 10 personnes pour 100 000, soit environ 4 000 à 6000 personnes en France.
La CSP touche majoritairement les hommes et elle affecte indifféremment tous les groupes ethniques.
Quels sont les symptômes ?
Le plus souvent, le diagnostic est posé avant même que des symptômes n’apparaissent. Le patient est asymptomatique et le diagnostic est posé devant des anomalies du bilan hépatique (fortuit ou en présence d’une maladie inflammatoire chronique de l’intestin).
Le diagnostic peut aussi être évoqué en présence de symptômes :
- Asthénie (fatigue)
- Prurit (démangeaisons)
- Douleurs de l’hypochondre droit (région supérieure de l’abdomen)
- Angiocholites à répétition (épisodes d’infection par des bactéries de l’intestin provoquant fièvre, douleurs et augmentation de la cholestase)
Des signes de mauvaise absorption digestive des graisses, comme une diarrhée avec selles grasses ou un amaigrissement, peuvent également être évocateurs d’une CSP.
La CSP peut être diagnostiquée tardivement, à un stade avancé, lors de la survenue d’une cirrhose.
Quelles sont les particularités de la CSP ?
La CSP est une maladie chronique. Elle évolue de manière très variable selon les individus. Cette évolution est généralement lente, s’étalant sur plusieurs années. L’une des principales complications de la CSP est la cirrhose. Celle-ci résulte d’un remplacement important des cellules du foie par du tissu fibreux. Le foie a alors des difficultés à assurer ses fonctions de stockage, de synthèse et de dégradation conduisant à une insuffisance hépatique.
Parmi les CSP, certaines formes se distinguent.
CSP des petits canaux biliaires
Anciennement appelée « péricholangite », cette forme, qui touche les petits canaux, représente environ 10 % des CSP. Certains cas peuvent s’étendre, après plusieurs années d’évolution, aux grands canaux biliaires. Considérée un temps comme un stade très précoce de la CSP, il semble plutôt s’agir d’une forme particulière de la maladie.
CSP à immunoglobulines de type G4
Cette forme très particulière et découverte récemment, est caractérisée par une augmentation souvent brutale de certains anticorps circulants dans le sang, les immunoglobulines de type G4 (IgG4). Son origine serait auto-immune, voire allergique. De ce fait, elle est parfois classée comme cholangite sclérosante secondaire.
Une hépatite auto-immune ou Overlap syndrome
L’association d’une CSP à une hépatite auto- immune, appelée syndrome de chevauchement CSP-HAI ou overlap syndrome, est plus fréquente chez l’enfant. Elle est caractérisée par la présence d’anticorps dirigés contre l’organisme.
Les conséquences de la CSP
Les conséquences liées à la cholestase peuvent être diverses :
- L’ictère ou la coloration des urines
- La stagnation de la bile favorise le passage dans le sang de certaines substances se trouvant dans le foie. Ainsi, la bilirubine issue de la dégradation de l’hémoglobine par le foie peut passer dans le sang. Sa couleur jaune prononcée peut provoquer un ictère (jaunisse) ou une coloration des urines.
- Une augmentation importante de la γ-GT et des PAL
- On retrouve aussi dans le sang certaines enzymes du foie telles que la γ-glutamyltransférase (γ-GT) et les phosphatases alcalines (PAL). Une augmentation anormale de ces enzymes dans le sang est déclenchée par la cholestase (réduction de la sécrétion de bile) et peut conduire au diagnostic de CSP. Ces enzymes sont également surveillées pour évaluer l’évolution de la maladie.
- Un prurit
- Par un mécanisme encore mal identifié, la cholestase peut provoquer des démangeaisons (prurit) parfois gênantes.
- La cholestase réduit l’absorption des graisses au niveau de l’intestin. Cela se traduit par :
- la présence de graisses dans les selles (stéatorrhée)
- un amaigrissement
- l’assimilation réduite des vitamines solubles dans les graisses (vitamines A, D, E et K). Il faut surveiller l’apparition d’éventuelles carences vitaminiques
- La CSP peut favoriser la formation de calculs biliaires (lithiase biliaire) qui peuvent augmenter la cholestase.
La CSP peut se présenter sous des formes très variées selon les individus et il n’est pas rare qu’elle soit associée à d’autres affections, en particulier les maladies inflammatoires chroniques de l’intestin (MICI).
En France, les études montrent une association dans environ 2 cas sur 3 avec, entre autres, une rectocolite hémorragique (RCH) ou une maladie de Crohn. La RCH concerne 83 à 99 % des cas de MICI associée et est d’ailleurs diagnostiquée avant même la CSP chez plus de 60 % des patients.
L’association avec une MICI augmente le risque de développer une cancer colo-rectal. Cela justifie une coloscopie de dépistage chaque année.
Comment fait-on le diagnostic d’une CSP ?
La CSP est souvent suspectée suite à une simple prise de sang révélant des résultats hépatiques anormaux. Toutefois, ces résultats hépatiques ne constituent qu’un signal d’alerte ou d’orientation car il n’existe pas de test sanguin permettant le diagnostic à lui seul.
Les examens complémentaires
Il est donc nécessaire de pratiquer des examens complémentaires pour mettre en évidence la présence d’anomalies typiques des voies biliaires :
- La bili-IRM, ou cholangio-IRM, est une IRM (Imagerie par Résonnance Magnétique) permettant de ne visualiser que les liquides biliaires et donc de déduire toute anomalie de leur écoulement, au niveau des canaux biliaires ou du pancréas. Il s’agit d’un examen essentiel pour poser le diagnostic de CSP, avec la mise en évidence de rétrécissements des canaux biliaires (sténose) associés à des dilatations. Toutefois, la lecture de ce type d’IRM est complexe et certaines formes particulières de CSP, comme celles qui touchent les petits canaux biliaires, peuvent nécessiter une biopsie du foie.
- La biopsie permet de prélever des fragments de tissus du foie afin de les étudier et de déterminer la présence d’inflammation et de fibrose. Elle n’est pas systématique lorsque la bili-IRM est déjà positive. Elle est réalisée à visée diagnostique pour rechercher des formes de CSP particulière comme le syndrome de chevauchement avec l’hépatite auto-immune (overlap syndrome) ou la CSP à petits canaux.
- L’élastométrie (Fibroscan) qui apprécie l’élasticité des tissus hépatiques et peut permettre d’évaluer l’existence de fibrose ainsi que suivre son évolution.
Evaluer le stade de la maladie
La biopsie du foie ou l’élastométrie permettent d’apprécier le stade de fibrose (stades I à IV). La biopsie évalue également le degré de raréfaction des petits canaux hépatiques (ductopénie). Mais elle est plus rarement faite aujourd’hui (voir « biopsie » page 18).
On peut évaluer si la maladie est évoluée (cirrhose) lorsqu’il existe des signes d’hypertension portale à l’échograhie ou à l’IRM.
Rechercher des maladies associées
Selon les résultats des examens et les symptômes présentés, d’autres explorations peuvent être nécessaires pour déterminer si d’autres pathologies sont associées à la CSP.
La coloscopie est toujours indiquée pour rechercher une RCH, souvent latente, et pour surveiller des modifications des tissus du côlon ou un cancer du côlon.
Les traitements de la cholangite sclérosante primitive
L’origine et le développement de la CSP ne sont pas totalement compris à ce jour. Les objectifs de la prise en charge sont donc de soulager les symptômes, de prévenir et traiter les complications de la maladie.
La cholestase a pour conséquence de diminuer la sécrétion des acides biliaires induisant une mauvaise absorption des nutriments, et particulièrement des graisses, par l’organisme. Le principe du traitement consiste donc à réduire l’inflammation et à compenser ce déficit hépatique. La prise quotidienne d’acide ursodésoxycholique (AUDC) est le traitement de base8
Les cholangites associées à une hépatite auto-immune et les cholangites à IgG4 bénéficient en complément d’un traitement visant à réduire l’activité immunitaire, comme les corticoïdes.
La transplantation hépatique
La greffe est généralement proposée dans les formes très évoluées de CSP ou si la qualité de vie est très altérée. Elle est indiquée en cas :
- d’ictère prolongé, avec un taux sanguin de bilirubine supérieur à 100 μmol/l,
- d’épisodes répétés d’angiocholite pour lesquels un traitement antibiotique se révèle insuffisamment efficace,
- de cirrhose avec hypertension portale.
Après la greffe, il existe un risque de récidive de la CSP et de poussée de RCH chez les patients préalablement atteints. Mais le pronostic reste globalement bon.
Comment suivre la CSP ?
Une surveillance de la CSP est nécessaire pour en évaluer l’évolution. Un suivi est généralement proposé 2 fois par an mais il peut être adapté au cas par cas en fonction des besoins.
- Tous les 6 mois :
- Évaluation de votre état de santé général par votre médecin
- Bilan sanguin :
- dosage des marqueurs sanguins du foie (bilirubine, enzymes hépatiques, électrophorèse des protides, numération formule sanguine, plaquettes, taux de prothrombine)
- dosage éventuel de l’antigène CA19/9 pour aider à dépister la survenue éventuelle d’une tumeur
- Une fois par an :
- Imagerie des voies biliaires :
- échographie pour étudier la vésicule biliaire en particulier
- IRM en cas de symptômes ou de modification des tests hépatiques
- Coloscopie :
- annuelle en cas de RCH
- tous les 5 ans en absence de RCH au début de l’évolution
- Élastométrie afin d’évaluer l’évolution de la fibrose du foie
- Imagerie des voies biliaires :
En cas de rétrécissement biliaire isolé ou évolutif, il peut être utile, après imagerie (IRM ou scanner), de réaliser un brossage endobiliaire (prélèvements de cellules au niveau de la sténose) en endoscopie ou abord percutané sous échographie pour déterminer la cause de ce rétrécissement.
Puis-je guérir de la CSP ?
La cholangite sclérosante primitive est une maladie chronique qu’on ne sait pas guérir dans l’état actuel des connaissances. Le traitement se prend à vie et vise à maintenir la maladie sous contrôle, pour continuer une vie aussi normale que possible.
Vais-je transmettre la maladie à mes enfants ?
Pour développer la maladie, on considère qu’une prédisposition génétique familiale est nécessaire, mais néanmoins non suffisante. Il ne s’agit en aucun cas d’une transmission inexorable comme pour les maladies purement génétiques (mucoviscidose, hémophilie, …).
Il n’y a pas de contre-indication à attendre un bébé quand on a une CSP, sauf à un stade très avancé.
Interview du Pr. Vincent Leroy sur Cholangite Sclérosante Primitive
(version longue 17mn)

Le livret Cholangite Sclérosante Primitive : comprendre et gérer son quotidien, publié par le laboratoire Mayoly Spindler et l’association albi vous sera peut-être remis par votre médecin, mais vous pouvez le télécharger ici en PDF.